
LAS ANOMALÍAS MÉDICAS MÁS
RARAS DEL MUNDO
15.
DIPROSOPUS O DIPROSOPIA:
Diprospus
(llamado en algunas ocasiones duplicación cráneo-facial) es un desorden muy
raro en el cual el afectado forma en su gestación dos caras; los niños con dos caras tienen un cuerpo y
extremidades normales, pero la mayoría de sus rasgos faciales están duplicados.
Según el grado de la alteración pueden tener dos, tres o
cuatro orejas; cuatro ojos (o sólo tres), y dos labios separados o uno sólo de
mayor tamaño q lo normal.

Los
niños con dos caras tienen un cuerpo y extremidades normales, pero la mayoría
de sus rasgos faciales están duplicados. Según el grado de la alteración pueden
tener dos, tres o cuatro orejas; cuatro ojos (o sólo tres), y dos labios
separados o uno sólo de mayor tamaño que lo normal; a pesar de lo que se puede
pensar no se produce como fusión o la división de dos embriones.

Existen
diversas teorías al respecto, una de las que más fuerza tiene nombra como
causante de esta anomalía a una proteína que determina la formación facial del
feto, cuando existe en exceso puede provocar una segunda cara y en su defecto
el rostro puede aparecer con algunas carencias; las personas que nacen con esta
anomalía suelen fallecer al poco tiempo de su alumbramiento, por suerte es una
afección muy fácilmente detectable en las primeras semanas de gestación.
14- INSENSIBILIDAD
CONGÉNITA AL DOLOR (La vida sin dolor)
Este
niño es Roberto Salazar, un niño de 5 años de apariencia perfectamente
normal; Roberto se muestra tan sonriente
y feliz como los demás, el problema es que si se golpeara con la cabeza contra
una pared, o se partiera un brazo por la mitad, seguiría jugando tan normal.
Es
incapaz de sentir dolor

Roberto
es una de las pocas personas en el mundo que sufre una enfermedad genética
conocida como CIPA o “insensibilidad congénita al dolor”.
Los
familiares de los enfermos de CIPA saben que el mundo sin dolor es un infierno.
Si no los vigilan de cerca, los niños se auto mutilan a la menor ocasión. A
Roberto tuvieron que sacarle todos los dientes de leche porque se arrancaba
trozos de su propia lengua. También se ha dañado las manos y le tienen que
alimentar a través de un tubo estomacal. Al no distinguir entre frío y calor,
una comida demasiado caliente le podría abrasar.
Como
consecuencia de esto, las personas que padecen esta enfermedad, suelen morir
más jóvenes por traumatismos y lesiones varias al no sentir ningún daño. Deben
estar bajo supervisión en edades tempranas para que no se lesionen ellos
mismos.
Afortunadamente
la CIPA es una enfermedad extremadamente rara y solo la sufren unos cuantos
centenares de personas en el mundo. Aun así, es un buen recordatorio de lo poco
que duraríamos en este mundo si no dispusiéramos de un mecanismo de aviso tan
complejo e intrigante como el dolor.
13- SÍNDROME
DE MOEBIUS (La vida sin Sonrisas)
Es
una enfermedad rara del desarrollo; caracterizado por parálisis facial de por
vida, la gente con el síndrome de Moebius no puede SONREÍR o fruncir, y no
puede mover a menudo sus ojos de lado a lado. En algunos casos, el síndrome
también se asocia con problemas físicos de otras partes del cuerpo.

El
síndrome de Moebius es extremadamente raro, 2 importantes nervios craneales, el
6º y 7, no están totalmente desarrollados en estos pacientes, estos nervios
controlan tanto el parpadeo y movimiento lateral de los ojos, como las
múltiples expresiones de la cara, por lo que causa parálisis facial y falta de
movimiento en los ojos. La falta de
expresión facial puede acompañarse de babeo, dificultades en el habla y
problemas de pronunciación.

A
menudo a las personas que padecen esta enfermedad, se les puede encontrar
durmiendo con los ojos abiertos. Tienen grandes dificultades en succionar,
tragar, hablar y cualquier actividad en la que estén implicados los músculos de
la cara. Muchos de los otros 12 nervios craneales se pueden afectar, incluyendo
tercero, quinto, octavos, noveno, onceavo, y el doceavo.
12. EL HOMBRE BURBUJA (Rara enfermedad)
Chandra
Winsu, sufre una rara enfermedad en la piel que le provoca grandes tumores que
se desarrollan por todo su cuerpo.
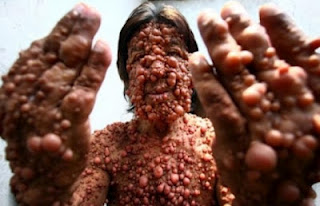
Estos
tumores comenzaron a desarrollarse en su cara cuando tenía tan solo 19 años,
con 24 se le había extendido a la espalda y con 32 ya tenía todo el cuerpo
cubierto de estas burbujas.
En
los inicios de la enfermedad, los padres del joven comenzaron un periplo de
médicos y dermatólogos sin demasiada fortuna. Todos quedaban asombrados y a la
vez desconcertados por los extraños síntomas.
Lo
único que sacaron en claro es que era genético y podía ser causado por una
anormalidad en el sistema nervioso que causa tumores benignos que se
desarrollan en la piel o en el hueso.
Cuando
se dio cuenta de que su hijo mayor Martín Ananda, de 32 años y su hija lis Candra
de 26 años desarrollaban la enfermedad decidió dar a conocer al mundo su
condición en un intento de encontrar una cura para sus hijos.
Estos
tumores son benignos y se desarrollan en
la piel y en los huesos.
11. MALDICIÓN DE ONDINA (Dormir puede costarte la vida)
(Hipo
ventilación alveolar primaria) Consecuencia de una o varias mutaciones del
genPHOX2B; de herencia autosómica dominante por lo que los mecanismos de la
respiración involuntaria no funcionan de manera adecuada y durante el sueño, la
persona afectada puede morir por falta de oxígeno.

Sin
embargo también existen personas que de alguna manera conviven con la enfermedad,
es decir que esta se presenta en forma leve, en donde el sujeto sigue viviendo
pero debido a que no puede dormir siente fatiga, dolores de cabeza, aumento del
nivel de glóbulos rojos entre otros.
En
general los pacientes tienen una ventilación adecuada despierta, pero
hiperventilan durante el sueño, de otro lado, la forma más grave de dicho mal
es cuando aparece desde el nacimiento y la mayoría de recién nacidos mueren sin
saber siquiera la causa.
10. EL SÍNDROME DE
ALICIA EN EL PAÍS DE LAS MARAVILLAS
Las personas con
síndrome de “Alicia en el País de las Maravillas” pasan por periodos de varios
días en que ven cómo un objeto se mueve solo o cómo su cuerpo parece aumentar o
disminuir de forma repentina.
Se trata de un trastorno bastante complicado y
que puede guardar alguna relación con las migrañas; quien sufre esta dolencia
es consciente de que sus visiones no son reales. Este síndrome resulta más
frecuente en niños o adolescentes, y también puede deberse a una enfermedad
infecciosa o una lesión cerebral; es una afección proclive a aparecer en
entornos familiares donde algún miembro ya ha experimentado este síndrome.

Según los expertos, las personas
afectadas por el síndrome de Alicia en el País de las Maravillas son en todo
momento conscientes de la naturaleza ilusoria de sus percepciones. Sin embargo,
éstas son lo suficientemente intensas como para que tengan que mirarse en un
espejo para comprobar su talla.
Aunque las pruebas diagnósticas aún no
han permitido identificar ningún área cerebral específicamente afectada, los
resultados de los estudios realizados en pacientes en su fase aguda mediante
tomografía computarizada revelan áreas de hipoperfusión en las proximidades del
tracto visual y córtex asociado, lo que podría explicar las quejas visuales de
los pacientes.
Los científicos sospechan que Charles
Lutwidge Dodgson, conocido bajo el pseudónimo de Lewis Carroll y afectado por
migrañas, pudo sufrir el síndrome, de forma que las experiencias de la joven
Alicia fueran bien conocidas por su creador.
9. SÍNDROME DE PROTEUS
El
síndrome de Proteus es extremadamente raro; desde que el Dr. Michael cohen lo
identificó en 1979, sólo se han confirmado poco más de 200 casos en todo el
mundo; puede que se hayan dado más, pero los que son correctamente
diagnosticados suelen tener manifestaciones evidentes del síndrome, estando
considerablemente desfigurados; hasta el momento no se le ha podido encontrar
el origen directo a este síndrome, aunque ninguna ha logrado ser confirmada,
pero al parecer todo indica que la condición radica en una mutación del gen
supresor tumoral PTEN homólogo de fosfatasa y tensina.

Es
una enfermedad progresiva, los niños suelen nacer sin ninguna deformidad
evidente; conforme crecen aparecen los tumores y el crecimiento de la piel y de
los huesos, la gravedad y la
localización de estos crecimientos asimétricos varían ampliamente, aunque
suelen darse en el cráneo, uno o más miembros y en la plantas de los pies.
Joseph
Carey Merrick nació en leicester, inglaterra el 5 de agosto de 1862 y falleció
en londres el 11 de abril de 1890. También conocido como “el hombre elefante”,
se hizo famoso debido a las terribles malformaciones que padeció desde los
dieciocho meses de edad. Condenado a pasar la mayor parte de su vida enrolado
en el mundo de la farándula, sólo encontró sosiego en sus últimos años de vida.
A
pesar de su desgraciada enfermedad sobresalió por su carácter dulce y educado,
así como por una inteligencia superior a la media y que sólo pudo demostrar en
sus postrimerías. Aunque todavía no se sabe con absoluta certeza, se cree que
joseph merrick pudo padecer una severa variación del síndrome de proteus, del
cual representaría el caso más grave conocido hasta el momento.

Hay
un riesgo de muerte prematura en los individuos afectados debido a trombosis y
trombo embolismo pulmonar, causadas por malformaciones asociadas a este
desorden en los vasos; otros riesgos
pueden venir provocados por el peso del tejido extra, se cree que Joseph Merrick
murió por el peso de su enorme y pesada cabeza que venció la resistencia de su
cuello y cayó hacia atrás, fracturándoselo.
8. ICTIOSIS
(Piel de
arlequín)
Trastorno
cutáneo hereditario en el que la piel aparece seca, como cuarteada, recubierta
de escamas de color más o menos oscuro, planas, a modo de escamas de peces. No
se trata de una sola enfermedad, sino de un grupo de trastornos, todos ellos
hereditarios y poco frecuentes, en los que existe una alteración de la
queratinización.

Podemos
incluir en la denominación de ICTIOSIS, diversos cuadros clínicos: la Ictiosis
simple, la Ictiosis ligada al sexo o Ictiosis X, la Ictiosis congénita grave,
el Collodion baby, la Ictiosis laminar, la Eritodermia congénita y otras formas
de malformaciones cutáneas ictiosiformes menos frecuentes.

La
ictiosis X se transmite por una herencia recesiva ligada al cromosoma X, por lo
que únicamente sufren la enfermedad los varones. Las lesiones cutáneas son más
extensas, afectando al cuero cabelludo y a la cara.
El
tratamiento de estas enfermedades es de tipo sintomático, a base de
medicamentos queratolíticos, que facilitan la exfoliación de las escamas
cutáneas, baños de sales, aplicación de urea localmente y la recomendación de
vivir en climas templados y con gran humedad ambiental.
7. ENFERMEDAD DE LEWANDOWSKY-LUTZ (Epidermodisplasia Verruciforme)
Es
una infección rara, permanente y grave por el Virus del Papiloma Humano que al
parecer es consecuencia de una deficiencia hereditaria en la inmunidad mediada
por células. Las lesiones pueden asemejarse a las verrugas planas, pero a
menudo son semejantes a la tiña versicolor y cubren el torso y las extremidades
superiores e inferiores. Se hizo popular gracias al documental de Discovery
Channel llamado "El Hombre Árbol".

La
enfermedad de Lewandowsky-Lutz es una epidermodisplasia verruciforme de
carácter autosómico recesivo con alteración de la inmunidad mediada por
células. Alrededor de 15 papillomavirus humanos están implicados en la
infección asociada, cuatro de los cuales son neoplasias cutáneas. La enfermedad
suele comenzar en la niñez con pápulas rojas y se disemina posteriormente por
el cuerpo como escamas grises o amarillas.
Uno
de los primeros análisis arrojó que la cantidad de glóbulos blancos –células
que combaten infecciones y virus- en la sangre de Dede eran tan bajos, que
Gasspari llegó a pensar que el asiático tenía VIH. Pero luego de profundizar
los estudios, el médico llegó a la conclusión de que la enfermedad del
"hombre árbol" se debía a un motivo mucho más extraño y misterioso.
Según Gasspari, Dede tenía una enfermedad genética que impedía a su sistema
inmunológico combatir a un virus que “secuestraba la maquinaria de las células
de la piel” y les ordenaba producir cantidades masivas de verrugas que le daban
ese aspecto parecido a las raíces de un árbol.

El
especialista informó que esa enfermedad se daba en “menos de una persona por
millón” y le propuso un tratamiento con dosis sintéticas de vitamina A –que
contrarresta el crecimiento de verrugas- para que luego de un tiempo el
asiático pueda volver a su aspecto normal.
“El
tratamiento durará de tres a seis meses. No va a quedar perfectamente normal,
pero las verrugas se deberían reducir en tamaño y cantidad lo suficiente como
para que pueda usar sus manos”, concluyó Gasspari, quien además reveló que
nunca había visto algo así en toda su carrera
6. PROGERIA (síndrome hutchinson-gilford)
Es
una enfermedad q comienza en la niñez y produce un envejecimiento rápido; la
progenia es una condición rara, pero ha llegado a ser de conocimiento público,
debido a sus síntomas q se asemejan bastante al envejecimiento humano normal y
a la aparición de varios niños afectados en las películas o televisión.
La
mayoría de los casos de progeria se producen por mutaciones de herencia
autonómica dominante en el gen LMNA, este gen participa en el mantenimiento de
la estabilidad nuclear y la organización de la cromatina. También podría
intervenir en la regulación de la expresión genética, la síntesis y reparación
del ADN.

La
Progenia produce un rápido envejecimiento de los niños, comenzando con una
deficiencia en el crecimiento durante el primer año de vida que tiene como
resultado cuerpos desproporcionadamente pequeños en relación con el tamaño de
sus cabezas.
Síntomas:
deficiencia en el crecimiento durante el primer año de vida, cara alargada y
arrugada, calvicie, pérdida de las pestañas y las cejas, estatura baja, cabeza
grande para el tamaño de la cara (macrocefalia), la fontanela (punto blando)
permanece abierta, mandíbula pequeña (micrognatia), piel seca, descamativa y
delgada, rango de movimiento limitado, retardo en la formación o ausencia de
los dientes.
La
causa de muerte generalmente está relacionada con el corazón o un accidente
cerebro - vascular como resultado de la aterosclerosis progresiva.
5. Porfiria
Eritropoyética Congénita o Síndrome de Gunther (EPC)
La
Porfiria se caracteriza por la emisión de orinas rojas, y una gran
sensibilización al sol, con erupciones bullosas o vesiculares sobre las zonas
del cuerpo descubiertas, que al repetirse ocasionan deformaciones y
mutilaciones. La Eritrodoncia.
(Coloración rosada de los dientes), es frecuente en las personas que padecen
esta enfermedad, así como también la Anemia Hemolítica.

Causas
Normalmente,
el cuerpo descompone químicos llamados porfirias en HEMO; las principales
fábricas de porfirinas del organismo son el hígado y la médula ósea.
En
esta médula ósea se sintetiza mucho HEMO para producir suficientes cantidades
de hemoglobina, sustancia tan necesaria para el trasporte de oxígeno; en
el hígado el HEMO se utiliza para formar otras substancias que fundamentalmente
sirven para desintoxicar.
Las
Porfirinas, que son desechos, salen del cuerpo a través de la orina o las
heces, pero las personas con porfiria tienen una anomalía genética que
interrumpe este proceso, como resultado, las porfirinas se acumulan en el
cuerpo, lo cual puede llevar a que se presenten erupciones en la piel,
sensibilidad a la luz, dolor abdominal, deformación de los huesos y otros
síntomas.
La
presencia de esta enfermedad en la familia aumenta el riesgo de padecerla,
proporcionalmente a la cercanía del parentesco. La enfermedad ataca con mayor
frecuencia a personas de raza blanca que a asiáticos y afro-descendientes.

Síntomas
Las
manifestaciones clínicas de las Porfiria pueden afectar a la piel (cutáneas) o
bien ser extracutáneas, o por otro lado de sintomatología mixta.
1. Síntomas cutáneos son la expresión de 2
fenómenos principales:
§ La Fotosensibilidad: con picor, escozor,
enrojecimiento, dolor e hinchazón en las zonas que son expuestas a la luz solar
durante unos minutos
§ Gran Fragilidad de la Piel: esta aparece de una
forma menos aguda y más progresiva con la aparición de ampollas bajo la
epidermis con pequeños roces; esto puede evolucionar a erosiones, cicatrices e
incluso Esclerodermia.
2.
Síntomas extracutáneos o sistémicos que son mucho más variables:
Afectación
hepática y del bazo; los
más característicos son los que se producen con el ataque agudo de porfirias.
Ésta se manifiesta como una explosión de síntomas digestivos, neurológicos y
psiquiátricos. Los síntomas digestivos incluyen náuseas, vómitos, dolor
abdominal.
Los
neurológicos implican neuropatía periférica con hormigueo, dolor muscular,
pérdida de fuerza, de forma simétrica en extremidades. Los trastornos
psiquiátricos incluyen confusión, alucinaciones, psicosis, etc.

Cuando
Bram Stoker creó el personaje de Drácula,
para su novela de terror publicada en 1897, lo hizo inspirándose en Vlad
Tepes, un singular príncipe de Valaquia que vivió en el siglo
XV.
Según
numerosas fuentes, ‘Vlad el empalador’ (como era conocido) padecía una
enfermedad llamada ‘PORFIRIA ERITROPOYÉTICA’ la cual se caracteriza, entre
otras cosas por:
ü retraer las encías
ü causar fotosensibilidad
(fotofobia)
ü anemia (en la que la
ingesta o contacto con el aroma que desprende un ajo puede agravarla)

La
exhaustiva documentación para crear al personaje de Drácula llevó a Stoker a
dotar al mismo de todos los síntomas de dicha patología, de ahí que los
vampiros que nos podemos encontrar en cualquier novela, cómic o film sean:
ü Sensibles a la luz solar
(debido a la fotofobia)
ü Necesiten sangre para
sobrevivir (a causa de la anemia, ya que los enfermos de porfiria
eritropoyética debían recibir transfusiones de sangre y cuando ésta aún no existía
era ingerida oralmente)
ü Les crecieran los
colmillos (por las encías retraídas que dejaban al descubierto mayor parte de
la dentadura)
ü Ser ahuyentados con una
ristra de ajos, que como se ha comentado
anteriormente, el comerlo u olerlo agrava severamente la enfermedad
El
tema de la estaca de madera y los crucifijos ya es cuestión de la ficción
literaria y nada tiene que ver con la patología que originalmente padecía el
personaje que inspiró la creación de Drácula y los posteriores vampiros nacidos
de la imaginación de otros autores.
La Porfiria Eritropoyetica Congenita, es una de las mas raras de
la “porfiria”, existen aproximadamente de 200 a 300 casos en el Mundo; las lesiones, provocan
dolorosas atrofias en la piel, que van destruyendo o mutilando el rostro o
cuerpo del afectado.
4. EPIDERMÓLISIS
BULLOSA
La
Epidermólisis Bullosa (EB) es una afectación congénita de la piel que se
caracteriza por la aparición de ampollas o vesículas con contenido de líquido
seroso-hemático. Al romperse dejan lesiones parecidas a quemaduras, de tamaño
variable y de cicatrización tórpida. Su evolución produce multitud de
cicatrices e importantes retracciones en la piel que provocan discapacidad
funcional, como la sindactília.
Las lesiones no se limitan a la piel, también
afectan a membranas y mucosas provocando complicaciones del tracto
gastrointestinal con dificultad nutricional (la malnutrición es un factor de
retraso en la cicatrización y de la calidad de las proteínas de anclaje), en la
que muchas veces se requerirá de alimentación enteral. No se recomienda el uso
de sonda nasogástrica por las potenciales lesiones que puede causar en las
mucosas. Se elige gastrostomía o dilatación gastroesofágica.

También
ocasionan un importante retraso en el desarrollo, problemas de visión y
dentales. Estos pacientes suelen presentar problemas hematológicos como anemia
severa que además de provocar un retraso en la cicatrización de las heridas,
provoca una descalcificación ósea localizada sobre todo en la columna
vertebral. Todo ello conlleva complicaciones severas que derivan en infecciones
secundarias y deterioro del estado general del paciente. Los enfermos, además,
tienen mayor probabilidad de presentar cáncer de piel antes de los 25 años.
La
EB puede variar desde un trastorno ligero a uno severo y debilitante y, algunas
veces, hasta a una enfermedad fatal. Las diferentes formas clínicas de EB
dependen del lugar donde hacen aparición las alteraciones estructurales y de la
afectación del nivel epidérmico. Y según esta afectación, del grado de EB,
serán las secuelas.

En
la forma distrófica la presentación es más severa y su pronóstico más grave. La
curación de las lesiones da lugar a cicatrices hipertróficas con deformaciones
en las extremidades y que pueden provocar estrechamiento de la vía
oro-faríngea. Aunque a grandes trazos estas son las tres presentaciones
principales, existen subtipos de la enfermedad que no se limitan sólo a estas
características tan evidentes.
La
EB es una enfermedad genética hereditaria. No es contagiosa ni infecciosa. Un
niño afectado mantiene la enfermedad y la forma en que se presenta toda su
vida. El diagnóstico y su clasificación no siempre resultan fáciles. La mayoría
de los diagnósticos más precisos se basan en la combinación de técnicas inmuno-histológicas:
microscopía electrónica, inmuno-fluorescencia directa y estudios con
anticuerpos monoclonales.
En
la actualidad es posible realizar un diagnóstico prenatal mediante biopsia de
la placenta a fetos de alto riesgo. El gen responsable de algunas de las formas
de EB se conoce desde 1986: una mutación en el gen 3p (21) que codifica para la
proteína del colágeno tipo VII, componente de las fibras de anclaje que forman
la unión dermo-epidérmica.
El
tratamiento para los afectados de EB se basa en unos buenos hábitos de higiene
y la importancia de la limpieza y tratamiento de las heridas. Asimismo, es muy
importante la prevención del mínimo traumatismo que haga perder la integridad
de la piel. El baño es básico para mantener la limpieza óptima de la superficie
de la piel y evitar, así, problemas de infección. Los productos usados deben
ser sumamente suaves e hidratantes, para prevenir la resecación cutánea.
3. COLA HUMANA VERDADERA (cola vestigial)
La
cola vestigial o cola humana verdadera, es una extravagancia fenotípica o un
atavismo de la que apenas se conocen unos 100 casos en el mundo; aunque las
causas no se conocen, todo hace suponer que se trata de “un fallo “en el
sistema celular que hace que se activen los genes que en tiempos pretéritos
dotaron de cola a nuestros ancestros.


Esos
genes que evolutivamente quedaron inactivos hace decenas de miles de años,
parecen ser que todavía están en algún lugar y que en contados casos, aparecen
para orgullo de los darwinistas; estas colas crecen en la zona final del sacro,
a la altura del cóccix y suele estar compuesta de tejido conectivo, músculos,
vasos sanguíneos, nervios y piel en la mayoría de ocasiones; y en otras pocas también
tiene vertebras y cartílago.
Se
suelen extirpar a los pocos días del nacimiento, pues no tiene ninguna
funcionalidad.
2. FIBRODISPLASIA OSIFICANTE
PROGRESIVA
Es
una extraña enfermedad genética que afecta al tejido conectivo, ligamentos,
tendones, fascias y cápsulas articulares; dicha enfermedad provoca la formación
de cartílago y hueso en los tejidos musculares en que se encuentra; es una
enfermedad genética, originada en un gen defectuoso que es transmitido por uno
o ambos padres, siendo suficiente que uno de ellos lo haga; de herencia
autosómica dominante; se piensa que
están implicados varios genes encargados de sintetizar factores de crecimiento
óseo; la enfermedad se manifiesta ya en la infancia, provocando brotes, donde
los tejidos blandos se inflaman y al desaparecer la inflamación, queda un nuevo
tejido óseo; de forma que las articulaciones se osifican de manera progresiva
hasta perder la movilidad.

El
causante de la enfermedad es una mutación en el gen del receptor de Activin
tipo IA
§ Activin receptor typeia
§ Bmp type I receptor acvr 1
La
fibrodisplacia comienza generalmente por el cuello, los hombros y la espina
dorsal, prosigue por los codos, cadera y rodillas; la enfermedad suele afectar
al tejido estriado y no al tejido liso; quedando libres de osificación los
músculos de la lengua, la cara, los ojos, la pared abdominal, el diafragma y el
corazón.
La
enfermedad puede diagnosticarse tempranamente por ciertas malformaciones
asociadas que ayudan a identificarla, como la microdactilia (dedos pequeños) y
la anquilosis en las falanges de los dedos gordos de los pies; la fibrodisplasia es progresiva, ocasionando
deformidades en la medida que avanza, hasta provocar incapacidad y trastornos
de conducta. la pérdida de la movilidad es grave y puede inmovilizar
completamente al paciente.
Los
síntomas comienzan a desencadenarse generalmente entre los tres y cinco años y
la evolución de la enfermedad es impredecible, presentando períodos de latencia
y de agravación; existen algunos factores que perjudican el desarrollo de la
enfermedad, como los traumatismos musculares, inyecciones intramusculares,
intervenciones quirúrgicas, tratamientos odontológicos que requieran del uso de
anestesia local, entre otros
La
cuarta parte de los pacientes desarrolla sordera y calvicie. Su diagnóstico se
realiza por evidencia clínica y radiológica, pues no se presentan indicios en
los análisis de laboratorio.
1. SÍNDROME HOMBRE LOBO (Hipertricosis Lanuginosa Congénita)
Es
una enfermedad extremadamente rara, ya que solo se han documentado 50 casos
desde la edad media; las personas que lo padecen están completamente cubiertas
por un vello lanugo largo, excepto en las palmas de las manos y de los pies.

El
laguno es el pelo fino (como si fuera pelusilla) que aparece en los recién
nacidos, en hombros y brazos; y que desaparece normalmente tras el primer mes
de nacimiento. En las personas que padecen esta forma de hipertricosis, el
laguno persiste y puede crecer durante toda la vida.
La
incidencia natural (sin contar los casos de las familias) se estima en un caso
entre 500 millones de habitantes; su causa es desconocida, se piensa qUE es una
mutación que sigue una herencia autonómica dominante; la mayoría es de herencia
familiar y muy raramente la mutación se da de forma espontanea.
(JIMMY PIT)

